Das CS-2-System von Cerebras Systems am Leibniz-Rechenzentrum unterstützt Bioinformatiker:innen dabei, die Codes von Proteinen zu entschlüsseln. Damit können neue Heilverfahren entstehen oder drängende Umweltfragen gelöst werden.
Proteine sind der Grundstoff des Lebens. Sie bestimmen Form, Aufbau und Funktionen von Zellen, Gewebe, Organen sowie den Stoffwechsel und das Wachstum, und zwar von Mensch, Tier, Pflanze. Ein Großteil von ihnen setzt sich aus etwa 20 Aminosäuren zusammen, so ein Ergebnis von rechnergestützten Analysen der letzten Jahrzehnte. Zwar sammeln sich inzwischen in vielen Datenbanken Milliarden von Aminosäure-Kombinationen und Variationen von Proteinsequenzen, doch wie diese Eiweißstoffe für Wachstum sorgen oder Zellfunktionen beeinflussen, ist noch immer weitestgehend ein Rätsel. Und: Die Suche in den prall gefüllten Datenbanken dauert viel zu lange. Doch Künstliche Intelligenz (KI) und Mustererkennung, neuerdings vor allem Modelle zur Verarbeitung menschlicher Sprache helfen, den Code des Lebens zu knacken. Sie beschleunigen zudem die Suche: „Sprachmodelle lernen Muster und Ähnlichkeiten in den Sequenzen direkt aus Proteindatenbanken“, erklärt Dr. Michael Heinzinger, Bioinformatiker der Technischen Universität München (TUM) und Mitarbeiter des Rost-Labs am Lehrstuhl Bioinformatik und numerische Biologie von Prof. Burkhard Rost. „Die traditionelle, statistische Mustererkennung dauert in der Regel lange und funktioniert nicht mit allen Proteinen gleich gut. Sprachmodelle verkürzen den Such- und Analyseprozess und geben uns neue Werkzeuge an die Hand, um das Verständnis von Proteinen zu verbessern.“
Sprachmodelle entschlüsseln den Protein-Code
Die Suche nach bestimmten Proteinstrukturen anhand von wiederkehrenden Mustern kann Tage, wenn nicht sogar Wochen dauern. Vor etwa vier Jahren entdeckte das Team um Prof. Burkhard Rost die Analogie von menschlicher Sprache und Proteinen: Die 20 wichtigsten Aminosäuren funktionieren wie Buchstaben, fügen sich quasi zu Wörtern und Sätzen, besser zu Proteinen und -Sequenzen mit eigenen Funktionen. Diese müssten, so eine Annahme am Rost-Lab, folglich mit smarten Programmen zur Verarbeitung natürlicher Sprache zu entschlüsseln sein. Statt wie gewohnt mit Artikeln von Wikipedia wurde daher ELMo, das Embeddings from Language Model, mit Proteinsequenzen gefüttert und trainiert. Das Ergebnis war SeqVec (Sequence-to-Vector), das erste smarte Modell zur Verarbeitung von Protein-Codes, das Datenbanken nicht mehr nach Mustern durchsucht, sondern direkt den Protein-Code aufnimmt und diesen eigenständig auf neue Proteine überträgt. Nach dessen Vorbild trainierten Forschende weltweit mit Large Language Models (LLM) und entwickelten bessere, effizientere Trainingsmodelle für Proteine mit neuen Funktionalitäten. Neue Technologien für die Verfahren der KI befeuerten diese Entwicklung.
Insbesondere die sogenannten Transformer – mit ihrer Hilfe wandelt der Computer Buchstaben- und Zeichenfolgen in mathematische Vektoren um – erweiterten die Möglichkeit des Trainings mit NLP-Modellen. Für die Protein-Analyse veröffentlichte der TUM-Lehrstuhl Bioinformatik verschiedene, auf Proteinsequenzen trainierte Transformer, namens ProtTrans. Diese ersten Proteincode-Modelle entstanden zunächst am eigenen Computercluster des Lehrstuhls sowie an den AI-Systemen des Leibniz-Rechenzentrums (LRZ), also auf parallel geschalteten Graphics Processing Units (GPU), speziell fürs maschinelle Lernen optimierte Prozessoren. Seit vergangenem Jahr steht den Wissenschaftler:innen am LRZ noch ein Cerebras CS-2 System zur Verfügung. „Ein völlig anderes KI-System“, sagt Heinzinger. „Mit seinem großen Chip und der hohen Speicherkapazität vereinfacht es viele Arbeitsschritte des verteilten Trainings. Wir müssen uns beispielsweise nicht mehr um die Kommunikation zwischen Prozessoren und Knoten kümmern.“ Modelle könnten schneller optimiert werden, nach ersten Erfahrungen von Heinzinger und seinen Kolleg:innen wird auch das Training mit großen Datenmengen beschleunigt. Allerdings fielen grundlegende Veränderungen oder Neuerungen an einem Modell vergleichsweise schwer. „Das ist nur eine Frage von Zeit, das CS-2-System erfordert für den Einsatz in der Forschung weitere Komponenten, und mit diesen kommt ein überarbeiteter Software Stack“, sagt Nicolay Hammer, promovierter Astrophysiker und Leiter des Teams Big Data & AI am LRZ.
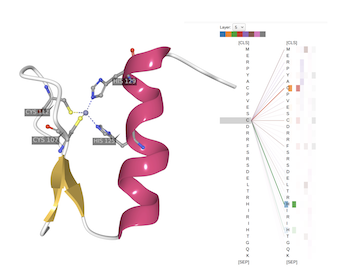
Die Grafik verdeutlicht die Funktionsweise von Sprachmodellen, die Kennungen für Aminosäuren wie Buchstaben zu Sequenzen oder Wörtern fügen
Ging es bislang vor allem ums Kennenlernen von Protein-Sequenzen, den Wörtern des Lebens-Codes, sollen die neuen, besseren und spezialisierten Sprachmodelle nun auch die Syntax und Grammatik des Protein-Codes dechiffrieren und aufzeigen, wie und wofür Eiweißstoffe dreidimensionale Strukturen bilden, sich falten und was das bewirkt. „Wir können mit größeren Sprachmodellen nicht nur die Proteine besser verstehen, sondern diese auch zielgerichtet manipulieren oder neu schreiben, um damit den Herausforderungen des 21. Jahrhunderts zu begegnen“, ergänzt Heinzinger Möglichkeiten. „Durch ihr breites Funktionsspektrum sind Proteine in vielen pharmakologischen und biotechnologischen Prozessen unentbehrlich.“ Mit ihnen werden zum Beispiel Medikamente hergestellt, neuerdings auch Biokraftstoffe oder Materialien, die Plastik abbauen oder Kohlenstoffe binden. Und wer den Code der Proteine beherrscht, kann damit unbegrenzt neue Moleküle oder Stoffe schaffen.
Rechenkraft verkürzt das Training
Sprach- und Proteinstrukturen ähneln sich, doch es gibt natürlich auch Unterschiede. Um Computern und smarter Sprachverarbeitung die Eigenheiten von Proteinen zu vermitteln, konfrontierten die Spezialist:innen des Rost-Labs erst ELMo und weitere Transformer-basierte Sprachmodelle mit Datensets, die zwischenzeitlich bis zu 2,3 Milliarden und mehr Proteinsequenzen enthalten. Normalerweise registrieren Sprachmodelle Buchstaben- und Wortkombinationen, in diesem Fall aber, wann sich welche Aminosäuren aneinanderreihen. Ähnlich einem Lückentext in der Schule füllen die Programme danach künstlich erzeugte Leerstellen und beweisen damit, dass sie den Aufbau der Proteine nachvollziehen können. Sucht das menschliche Hirn mit Intuition und nach Sinn fehlenden Wörtern, sortiert die Maschine Lösungen nach statistischer Verteilung. Sie nennt verschiedene Variationen für die Lücke nach den Wahrscheinlichkeiten, mit der diese in Proteinen auftreten. „Je besser Computer eine Proteinsequenz lesen können, umso besser können sie deren Strukturen und Funktionen verstehen“, so Heinzinger. Schrittweise werden danach Parameter, die Kriterien für Auswahl und Analyse, verändert oder hinzugefügt.
Mehrere Dutzend Trainingsläufe sind die Regel. Mit jedem Parameter wachsen zudem die Modelle und neuronalen Netzwerke: Enthielt SeqVec 93 Millionen Neuronen, enthält das jüngste Modell aus dem Rost-Lab ProtT5 schon drei Milliarden: „Ein Sprachmodell zu trainieren, ist teuer, es dauert mit jedem Durchlauf länger, weil die Datensätze, aber auch die Zahl der Parameter wachsen. Das braucht Rechenkraft und Energie“, sagt Bioinformatiker Heinzinger. „Aber danach bekommen Anwender:innen aus Biotechnologie, Pharmakologie oder Medizin im besten Fall ein brauchbares Modell für ihre eigenen Analysen an die Hand.“
Je nach verfügbarer Rechenleistung und Größe der Modelle dauern Trainingsläufe Stunden oder mehrere Wochen. Dabei werden Daten immer wieder durch Prozessoren und Speichereinheiten geschleust, neu kombiniert, ausgewertet, überprüft, abgespeichert, weiterverarbeitet. Stehen im LRZ AI-Cluster 68 parallel geschaltete GPU mit dynamischem Arbeitsspeicher (DRAM) von jeweils zwischen 16 und 80 Gigabyte zur Verfügung, bietet das Cerebras-System einen einzigen Chip, auf dem sich insgesamt 850.000 Arbeitseinheiten rund 40 Gigabyte Speicher teilen. Dieser ist auch noch mit HPE Superdome Flex Servern verbunden, die weitere 10 Terabyte Arbeits- (RAM) sowie 100 Terabyte Datenspeicher mitbringen. So kommt eine Rechenleistung zusammen, für die sonst Dutzende von GPU erforderlich wären. Daten können auf diesem Supercomputer blitzschnell zwischen Arbeits- und Speichereinheiten fließen. „Ein Training mit ProtT5 wäre auf den anderen LRZ AI-Systems nicht mehr möglich, es würde Jahre dauern“, sagt Heinzinger. „Im Gegensatz zur Mustererkennung, die für jedes neue Protein eine Datenbank immer wieder durchsuchen müsste, trainieren Sprachmodelle direkt auf den Rohdaten einer Datenbank. Die so aufgenommenen Proteincodes können danach direkt auf neue, andere Proteine angewandt werden.“
Zurzeit versucht der Bioinformatiker, unterschiedliche, etablierte LLM auf dem Cerebras CS-2-System zu implementieren und deren Tauglichkeit für Fragen aus Biologie und anderen Lebenswissenschaften zu überprüfen. Es geht dabei zunächst aber weniger darum, Proteine zu analysieren als die neue Technologie in den Griff zu bekommen und das System auf eigene Ansprüche anzupassen.
„Sprachmodelle eröffnen neue Möglichkeiten, etwa die Generierung von Texten oder Proteinsequenzen“, so Heinzinger. Spezialist:innen können nicht nur neue Stoffe, Materialien oder Medikamente mit den dechiffrierten Proteinsequenzen aufbauen, einmal austrainiert bieten Modelle wie ProtT5 außerdem Grundlagen für die Entwicklung von Software: etwa zur Analyse von Organismen und Gewebe, zum Erkennen und Behandeln von Mutationen oder Krankheiten, auch zur Entwicklung und Gestaltung neuer organischer Stoffe und Materialien. Auch dafür lohnt sich der hohe Aufwand des Trainings. (vs)

Dr. Michael Heinzinger., Bioinformatiker und Mitarbeiter am Rost-Lab/TUM